Ependymom (pathology)

Ependymomas (ICD 9391/3) are tumors histogenetically derived from the glia representing about 5% of all gliomas. They occur in the ventricles and spinal cord. pendymomas can establish implantation metastases through cerebrospinal fluid, anaplastic ependymoma can, exceptionally, also establish metastases outside the CNS. In general, the prognosis of ependymomas is not very good.
Tumor localization
Intracranial incidence is more common in childhood, and spinal cord ependymomas are more common in adulthood. At least two-thirds of children's tumors are located in the fourth ventricle and may thus be clinically manifested by an increase in intracranial pressure, which leads to obstructive hydrocephalus. When they are supratentorially located, they can cause epileptic seizures or motor deficits. In adulthood, 60–70% of cases are located in the spinal cord, mostly in the conus terminalis, filum terminale or cauda equina.
Macroscopic appearance
Ependymomas are usually greyish white, granular and friable in cross section. They are well defined in a lobular shape. Brain ependymomas are often cystically altered and foci of calcification may be present. In the case of intramedullary growth, fusiform thickening of the affected spinal cord segment occurs.
Histological image
Ependymomas can be heterogeneous, so a tumor can have a different appearance in different areas. The cytoplasm is usually abundant, with tumor cells sometimes having an astrocytic or gemistocytic appearance. The nuclei of low-grade pendymomas are monomorphic, round to oval, their chromatin is finely dispersed. The nuclei of high-grade lesions are polymorphic, irregularly shaped, and hyperchromic.
The basic histological characteristics of the ependymoma are non-infiltrative solid growth pattern, perivascular pseudorosets and ependymal rosettes.
Perivascular pseudorosets
Perivascular pseudorosets (syn. Perivascular rosettes) are non-nuclear zones around small blood vessels. The morphological basis of the rosettes are the radially oriented protrusions of the cells that surround the vessel. If the tumor is low in cell, the perivascular rosettes are poorly visible.
Perivascular pseudorosets occur not only in ependymomas, but can also occur in medulloblastomas, PNET (Primitive neuroectodermal tumors), central neurocystoma and glioblastomas.
Ependymal rosettes
Ependymal rosettes are concentrically oriented clusters of cells around an empty lumen. If the rosettes are larger, they may be difficult to distinguish from the normal lining of the ventricular system. Conversely, extremely small rosettes can be formed by only one cell as a dense intracytoplasmic eosinophilic nucleus, which is based on the accumulation of microvilli; this nucleus is positive for immunochemical staining for EMA (Epithelial Membrane Antigen).
Ependymal rosettes can be observed practically only in ependymomas, unfortunately they are rarely detected. This means that the presence of an ependymal rosette makes the diagnosis of ependymoma practically certain, the absence does not by far exclude the diagnosis of ependymoma.
Histological variants
Cell ependymoma
Cell ependymoma (ICD 9391/3) is the most common type of ependymoma. he tumor is formed by a dense network of cytoplasmic protrusions. These protrusions form collar-like densities around the vessels passing through the tumor, which are referred to as perivascular pseudorodes. Sometimes there may be formations of canals, tubules, or actual rosettes bounded by cells that look like normal ependymal cells. Less visible longitudinal grooving of the cores is usually described. Dystrophic calcifications are a common finding, and sometimes osseous or chondroid metaplasia can occur. Deposites of necrosis re relatively common. Nuclear pleomorphism may also occur, but this is not a sign of anaplasia.
Tanycytic ependymoma
Tanycytic ependymoma (ICD 9391/3) is an uncommon variant. Its cells are rather elongated, growing predominantly in bundles, and perivascular pseudorodes are poorly developed. Particularly poorly visible pseudoribes are the reason for the risk of confusing this variant with pilocytic astrocytoma and schwannoma.
The clear cell ependymoma
The clear cell ependymoma (ICD 9391/3) contains a more pronounced admixture of astrocytes, in which perinuclear clearing is evident. Especially if plexiform vascularization and spherical small calcifications (calcosferules) are present, this variant may be confused with oligodendroglioma. Perivascular rosettes can be quite inconspicuous and can only be elucidated by immunochemical staining on GFAP (Glial Fibrillary Acidic Protein). Endothelial proliferation may be detected in the tumor, which is a sign of poorer biological behavior.
Ring cell ependymoma
In some cases, intracytoplasmic lumens may form, so that ependymal cells become a sealing ring.
Papillary ependymoma
Papillary ependymoma (ICD 9393/3) is a relatively rare variant, it can be confused with tumors arising from the chorioid plexus or metastases of papillary carcinoma. It consists of one or more layers of cubic to cylindrical cells arranged around a central finger-like glial stroma.
Anaplastic ependymoma
Anaplastic ependymoma (ICD 9392/3) is characterized by poor differentiation, conspicuous cellularity and marked mitotic activity. Nuclear hyperchromasia is evident, nuclear pleomorphism is common. Microvascular proliferation is abundant.
Anaplastic ependymoma behaves aggressively, it can spread metastatically through cerebrospinal fluid. Exceptionally, metastases outside the central nervous system in the lymph nodes and lungs have also been described. Metastatic spread through the ventriculoperitoneal shunt into the abdominal cavity has also been described.
Myxopapillary ependymoma
Myxopapillary ependymoma (ICD 9394/1) ccurs almost exclusively in the conus medullaris and in the filum terminal, it is more common in younger adults. Myxopapillary ependymoma can spread through the cerebrospinal fluid , čmore often in pediatric patients. Clinically, it usually manifests as long-term back pain.
The tumor is highly vascularized, may be circulating, and is gelatinous in incision. The tumor is made up of cuboid cells, which are attached to the basophilic mucinone material, which surrounds the blood vessels and creates microcystic spaces. Degenerative stroma changes such as abundant collagen, vascular sclerosis, trombosis or bleeding are common. Myxopapillary ependymus is completely benign, only local recurrences are described. The possible presence of cellular atypia does not worsen the prognosis.
Subependymoma
Subependymoma (ICD 9383/1) differs from the above ependymomas. It arises from neurogli in the wall of the ventricles, nmost often in the fourth ventricle. There are also multifocal variants, but even in this case the tumor is benign. Subependymoma is usually an accidental autopsy, the clinical manifestation is exceptional, usually in adult patients.
Macroscopically, a subependymoma is usually a well-defined lobular solid formation pressing against the ventricular wall or slightly prominent. The cut is white, calcification is common, which is sometimes considerable.
Histologically, clusters are characterized by a small number of small tumor cells that are randomly scattered in a poorly vascularized fibrillar network. The net can have a spongy appearance. It is not an unusual formation of cysts. The nucleus of the tumor cells is oval with finely dotted chromatin. hyaline changes, can occur in blood vessels passing through the tumor as a result of hemorrhages an trombosis, leading to necrotization in the tumor.
Histochemical properties
Practically, always positive are:
- GFAP (Glial Fibrillary Acidic Protein) – negativity virtually rules out the diagnosis of ependymoma;
- NCAM (CD56) – diff.dg. choroid plexus lesions that are negative;
- vimentin;
- S-100;
- keratin AE1/AE3.
Positive can be:
- EMA (Epithelial Membrane Antigen);
- keratine CK7, CAM 5.2, CK903 a CK20.
Never positive:
- CEA (Carcino Embryonic Antigen);
- most neuronal antigens.
Proliferative antigen Ki67 shows higher activity in grade II and III tumors.
The Olig2 antigen has a special character . This can be positive for at most several cell nuclei; if its positivity is more pronounced, it practically precludes the diagnosis of ependymoma.
Grading
The WHO grading of ependymomas by histological type is as follows:
- grade I – myxopapillary ependymom, subependymom;
- grade II – cell ependymoma, papillary ependymoma, tanycytic ependymoma, clear cell ependymoma, RELA fusion-positive ependymoma;
- grade III – anaplastic ependymoma, ependymoma of clear cells (if endothelial proliferation and numerous mitoses are detected).
Diferential diagnostics
Diagnosis is usually easy, differential diagnostic embarrassment is exceptional.
Monomorphic angiocentric glioma
Monomorphic angiocentric glioma is a cortical lesion that was first described in patients with epilepsy. Like the ependymoma, it forms perivascular pseudorodes and shows the same pattern in immunochemical staining on EMA. The distinctly infiltrative character and the presence of elongated tumor cells in the subpial region serve to distinguish them.
Astroblastoma
Astroblastoma is a rare, well-defined glioma. It forms astroblastic rosettes around the vessels, incl. the same pattern of EMA positivity. The finding of perivascular fibrosis and hyalinization will be used to distinguish it from the ependymoma.
Papillary glioneural tumor
The papillary glioneural tumor is a supratentorial lesion, and the histological picture shows the formation of structures centered on hyalinized vessels surrounded by a GFAP positive fibrillar network.
Oligodendroglioma
Oligodendroglioma may histologically resemble a clear cell ependymoma. EMA and GFAP staining is used for resolution. A 1p / 19q deletion is evident in adult oligodendrogliomas.
Central neurocytoma
Central neurocytoma (formerly eponymous foramen Monroi). Histologically, it may resemble a clear cell ependymoma, but it is almost never immunochemically stained for GFAP. Characteristically, it usually grows from the septum pellucidum.
Pilocytic astrocytoma
Pilocytic astrocytoma can cause diagnostic difficulties with basic staining if the microcystic structure is lost. Immunochemical staining for Olig2 is of great diagnostic value, where in the case of pilocytic astrocytoma, all nuclei are stained.
Pilocytic astrocytoma
Pilocytic astrocytoma is the biggest diagnostic problem. Immunochemistry will be used to distinguish. The papillary tumor of the pineal gland is usually negative when stained for GFAPα and is significantly positive when stained for cytokeratins.
Paraganglioma
Paraganglioma can be a source of diagnostic difficulties in some locations.
Therapy
The most important prognostic factor is the extent of the surgical procedure. The basis of therapy is therefore as radical resection of the tumor as possible.
In the case of pediatric posterior tumor tumors, a moderate suboccipital approach is usually chosen. The cranial nerves are usually relatively close to the tumor, increasing the risk of damage. A general complication of posterior pit surgery, which can occur in this case as well, is syndrome of the posterior cranial fossa. An external ventricular drain, less commonly a ventriculoperitoneal shunt, is usually used as a hydrocephalus solution.
Access to tumors located in the spinal cord takes place via a standard laminectomy (= oan operation in which the posterior arch of one or more vertebrae is removed). Perioperative examination of somatosensory evoked potentials does not greatly affect the extent of the resulting neurological deficit, because the size of the resection is governed primarily by the size of the tumor, the experience of the surgeon and possibly perioperative biopsy.
The role of adjuvant chemotherapy or radiotherapy is still the subject of expert debate.
Prognosis
A significant prognostic factor is the radicality of the surgical procedure. Patients who underwent complete resection remained relapsed in 70% for 5 years, while with incomplete resection, only 30-40% of patients relapsed after 5 years. The overall five-year survival of patients with ependymoma is 50%, but in the case of ependymoma in the posterior fossa, the five-year survival is much worse. In grade II patients, about a quarter of patients die within seven years of surgical removal of the tumor, and more than two-thirds in grade III patients.
Image gallery
- Macroscopic appearance
-
 Myxopapillary ependymom filum terminale.
Myxopapillary ependymom filum terminale. -
 Ependymoma in the back pit of the skull.
Ependymoma in the back pit of the skull. -
 Macroscopic section, ependymoma IV. chambers.
Macroscopic section, ependymoma IV. chambers. -
 Macroscopic section, myxopapillary ependymoma of the spinal cord.
Macroscopic section, myxopapillary ependymoma of the spinal cord.




- Microscopic appearance
-
 Cell ependymoma (grade II), H&E. The image shows well-formed epithelial rosettes as well as perivascular pseudorodes.
Cell ependymoma (grade II), H&E. The image shows well-formed epithelial rosettes as well as perivascular pseudorodes. -
 Cell ependymoma (grade II), H&E. Detail.
Cell ependymoma (grade II), H&E. Detail. -
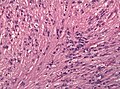 Tanicytic ependymoma (grade II), H&E. The image is dominated by elongated cells growing in the indicated bundles.
Tanicytic ependymoma (grade II), H&E. The image is dominated by elongated cells growing in the indicated bundles. -
 Clear cell ependymoma (grade II), H&E. Noticeable perinuclear clearing of the cytoplasm.
Clear cell ependymoma (grade II), H&E. Noticeable perinuclear clearing of the cytoplasm. -
 Papillary ependymoma (grade II), H&E. Visible papillary formations.
Papillary ependymoma (grade II), H&E. Visible papillary formations. -
 Anaplastic ependymoma (grade III), H&E. Nuclear pleomorphism and numerous mitoses are evident.
Anaplastic ependymoma (grade III), H&E. Nuclear pleomorphism and numerous mitoses are evident. -
 Myxopapillary ependymoma (grade I), H&E.
Myxopapillary ependymoma (grade I), H&E. -
 Detail jader ependymomu, H&E.
Detail jader ependymomu, H&E. -
 Subependymoma (grade I), H&E. A fibrillar network is evident, in which small clusters of tumor cells are scattered.
Subependymoma (grade I), H&E. A fibrillar network is evident, in which small clusters of tumor cells are scattered. -
 Subependymoma (grade I), H&E. Higher magnification.
Subependymoma (grade I), H&E. Higher magnification.










- Immunochemical staining
-
 GFAP staining.
GFAP staining. -
 EMA staining. Ependymomas are characterized by a dotted pattern of positivity.
EMA staining. Ependymomas are characterized by a dotted pattern of positivity. -
 EMA staining. A more pronounced periluminal positivity is evident.
EMA staining. A more pronounced periluminal positivity is evident. -
 EMA staining. Distribution of positivity in papillary ependymoma.
EMA staining. Distribution of positivity in papillary ependymoma. -
 Proliferation of Ki67 antigen in anaplastic appendix.
Proliferation of Ki67 antigen in anaplastic appendix.





Links
Related articles
- WHO classification of central nervous system tumors
- Ependymoma
- Astrocytoma
- CNS tumors (pediatrics)
- Spinal tumors
Literature
- ROSAI, Juan, et al. Ackerman's Surgical Pathology. 8. edition. St. Louis, MO : Mosby, 1996. 2; ISBN 0-8016-7004-7.
- WHO. . Mezinárodní klasifikace nemocí pro onkologii : česká verze. 3. edition. Praha : Ústav zdravotnických informací a statistiky, 2004. ISBN 80-7280-373-5.
- VEGE, KD – GIANNINI, C – SCHEITHAUER,. The immunophenotype of ependymomas. Appl Immunohistochem Mol Morphol. 2000, y. 8, vol. 1, p. 25-31, ISSN 1533-4058.
- GODFRAIND, C. Classifications and controversies in pathology of ependymomas. Childs Nerv Syst. 2009, y. 25, vol. 10, p. 1185-1193, ISSN 1433-0350.
External links
- BENEŠ, Jiří. Studijní materiály [online]. ©2007. [cit. 2009]. <http://www.jirben.wz.cz/>.
- BRUCE, Jeffrey N. Medscape : Ependymoma [online]. ©2013. [cit. 2014]. <https://emedicine.medscape.com/article/277621-overview>.
- FULLER, Christine. Medscape : Ependymoma Pathology [online]. ©2012. [cit. 2014]. <https://emedicine.medscape.com/article/1744030-overview>.
- PathologyOutlines.com. CNS tumors > Ependymal tumors : Ependymoma [online]. ©2012. [cit. 2014]. <http://www.pathologyoutlines.com/topic/cnstumorependymoma.html>.
- BUCHVALD, P – SUCHOMEL, P – KAISER, M. Chirurgická léčba ependymomů krční a horní hrudní míchy. Cesk Slov Neurol [online]. 2007, y. 70/103, vol. 2, p. 196-200, Available from <https://www.prolekare.cz/specialist-agreement>. ISSN 1802-4041.
- Liga proti rakovině Brno. Co potřebujete vědět o nádorech mozku [online]. [cit. 2014]. <http://www.onko.cz/_pub/publikace/mozek.pdf>.
