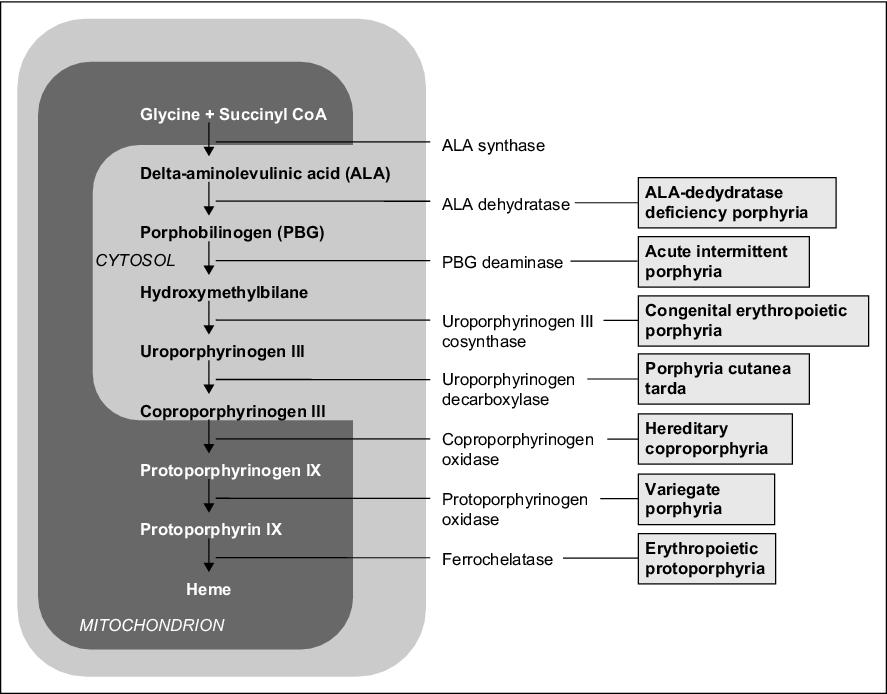
Porphyria

Porphyrias are a group of metabolic disorders of heme synthesis, characterized by the accumulation or increased excretion of porphyrins. Porphyrias can be erythropoietic, hepatic, or erythro-hepatic. The most common disorder belonging to this group of disorders is porphyria cutanea tarda.[1]
Porphyrins
Porphyrins are cyclic compounds consisting of four pyrrole rings joined by methine bridges (= CH-). In porphyrinogens, these bridges are methylene bridges (-CH2-). They form complexes with metal ions (e.g., Fe in hemoglobin, myoglobin, cytochromes, catalase; Co in vitamin B12; Mg in chlorophyll).
They are colored in various shades of red (unlike colorless porphyrinogens). Their color is due to the system of conjugated double bonds in their molecule, which absorb visible light. They fluoresce red in UV light. Due to their indispensable role for life, they must be de novo synthesized in the body (when ingested in food, they decompose and serve as a source of nutrients).
Characteristics of porphyrias
Mutations in the genes that control the synthesis of enzymes involved in heme synthesis lead to product deficiency just after the block or substrate accumulation just before the block.
In the case of an enzyme defect in the early phase of biosynthesis (before the formation of porphyrinogens), the starting substances ALA - δ-aminolaevulinic acid (5-aminolevulinic acid) and PBG (porphobilinogen) accumulate in body fluids, which has a toxic effect (interference with synaptic functions) on the PNS and CNS, which leads to characteristic symptoms - neuropathy (muscle weakness), abdominal pain, increased sympathetic activity, and neuropsychic problems (restlessness, hysteria, psychotic states).
Defects in later stages of synthesis lead to the accumulation of porphyrinogens, whose oxidation products (corresponding porphyrins) cause photosensitivity (hyperreaction to visible light in the 400 nm region). When porphyrins are exposed to light of this wavelength, they are excited, reacting with molecular oxygen to form oxygen radicals, which damage cellular organelles, including lysosomes → lysosomal enzymes are released, which damage light-exposed skin (erythema, blisters, scarring). Symptoms of the disease also include redness or browning of the teeth, urine, and stool, and receding gums.
Individual methods of elimination are important in the diagnosis of porphyria. ALA, PBG, and uroporphyrin are soluble in water → excreted in the urine. Protoporphyrin is insoluble in water → excreted in the bile. Coproporphyrin is found in both bile and urine, and its urinary excretion increases with liver damage.
Manifestation is often only in adulthood after a certain evoking moment (drugs, sun exposure, stress, hormonal effects). The administration of some drugs (barbiturates, anesthetics) as well as alcohol consumption increases the activity of ALA-synthase, which leads to increased production of porphyrins, and thus worsening of symptoms in patients with porphyria (possibly acute attack). Acute porphyrias more often affect women, while chronic forms more often affect men.
In acute therapy, glucose suppresses the induction of ALA synthase, the effect being potentiated by the application of heme derivatives such as heme-albumin, heme-arginate, or hematin. Heme inhibits ALA-synthase via negative feedback.[2]
Porphyrias can be classified in several ways:
- Affected porphyrin-producing cells:
- hepatic - AIP, PCT, HCP, VP, ADP
- erythropoietic - CEP
- erythro-hepatic - EPP
- Manifestations:
- cutaneous - PCT, EPP, CEP, but also VP and HCP
- liver - AIP, ADP, VP, HCP;
- course:
- acute - AIP, VP, HCP, ADP;
- chronic - PCT, EPP, CEP
| Porphyria type | Affected enzyme | The main symptoms | Laboratory finding |
| Acute intermittent (hepatic) | uroporphyrinogen I cosynthetase | abdominal pain, neuropsychiatric symptoms, no photosensitivity | U-PBG ↑,
U-uroporfyrin ↑ |
| Congenital erythropoietic | uroporphyrinogen III cosynthetase | photosensitivity | U-uroporfyrin ↑,
U-PBG ↓ |
| Porphyria cutanea tarda (cutaneous) | uroporphyrinogen decarboxylase | photosensitivity | U-uroporfyrin ↑ |
| Porphyria variegata (hepatic) | protoporphyrinogen oxidase | photosensitivity, abdominal pain, neuropsychiatric symptoms | U-PBG ↑,
F-protoporfyrin ↑ |
| Protoporphyria (erythro-hepatic) | ferrochelatase | photosensitivity | F-protoporfyrin ↑,
Ery-protoporfyrin ↑ |
| Hereditary coproporphyria (hepatic) | coproporphyrinogen oxidase | photosensitivity, abdominal pain, neuropsychiatric symptoms | U-PBG ↑,
U-uroporfyrin ↑ |
Cutaneous porphyria
Porphyria cutanea tarda (PCT)
It is an AD hereditary defect of uroporphyrinogen decarboxylase, with an incidence of 1:25 000 (the most common porphyria), especially in middle-aged men. Porphyrins are produced in excess in the liver, where they accumulate and then are transported through the bloodstream to the skin, where they cause photosensitivity, which is a typical symptom. After exposing the skin to sunlight, large blisters filled with fluid appear, which heal very slowly with the formation of scars and mildew (dotted whitish deposits). The skin is hyperpigmented, later atrophic, and vulnerable. Hypertrichosis occurs on the temples and around the eyes. The clinical manifestation is associated with liver damage caused by alcohol, polyhalogenated hydrocarbons (hexachlorobenzene, dioxin), estrogen treatment, hepatomas, hemochromatosis,or hepatitis. If left untreated, it can lead to liver cancer. There is also a non-hereditary form (sporadic, so-called PCT type 1). In the urine we find uroporphyrin, high levels of iron, and in 50% of cases high levels of liver enzymes.
Treatment: repeated venipunctures (300-500 ml at 2-4 week intervals) depriving the body of excess porphyrins and iron + administration of antimalarial chloroquine (125-250 mg daily), which causes slow leaching of porphyrins, protection from the sun (clothing, special creams), and a liver diet.
Congenital erythropoietic porphyria (CEP, Günther's disease)
This is an AR hereditary defect of uroporphyrinogen-III-synthase (UROS) leading to increased production of porphyrins in the bone marrow, which accumulate in the body, especially in erythrocytes. The incidence is 1: 2-3 million. This disease usually manifests itself in childhood. Manifestations of the disease vary - these include dark red urine (due to the presence of uroporphyrin and coproporphyrin), skin sensitivity (blistering, scarring) and darkening, eye sensitivity, eyelash loss, anemia, splenomegaly, red teeth, and excessive hair growth (especially on the hands and face).
Treatment: bone marrow transplantation, sun protection, blood transfusion, splenectomy
.jpg)
Erythropoietic protoporphyria (EPP)
This is an AD hereditary defect of ferrochelatase, which results in the accumulation of protoporphyrin in the liver, bone marrow, and skin. The most common symptoms are redness, itching and swelling of the skin even after a short (several minutes) exposure of the skin to sunlight. The symptoms disappear after hours to days, with repeated exposure leading to scarring of the skin and other variable skin manifestations. The disease usually manifests itself in childhood. In a small amount of cases, liver damage occurs.
Treatment: alleviation of symptoms with beta-carotene, antihistamines, melanotan, phototherapy, prevention is through the use of protective clothing and special creams. Unlike acute liver porphyrias, erythropoietic porphyrias are not exacerbated by any drugs.
Liver porphyria
Acute intermittent porphyria (AIP)
This is an AD hereditary defect in hydroxymethylbilane synthase (porphobilinogen deaminase, PBGD or uroporphyrinogen-I-synthase) leading to the accumulation of heme precursors in the liver. It manifests as an acute attack after exposure to certain chemicals (steroids, drugs, alcohol), starvation, infections, or stress; mostly during puberty. The main symptoms are abdominal pain (imitating acute abdomen), constipation, vomiting, hypertension, and mental problems (hysteria), headaches, paresis and plegia. There is an increased level of ALA and PBG in the urine. Hyponatremia, hypokalemia, and abnormalities in the metabolism of sugars and fats are evident. The diagnosis is confirmed by decreased PBGD activity in erythrocytes.
Treatment: in the acute phase, infusion with glucose (inhibits ALA-synthase) and hematin; prevention of another attack is the avoidance of the inducing substance (certain drugs or alcohol).
Porphyria due to a 5-aminolevulate dehydratase deficiency (ADP, Doss porphyria)
It is caused by an AR hereditary deficit of 5-aminolevulate dehydratase.
Symptoms include abdominal pain and neuropsychiatric problems. ALA and coproporphyrin are present in the urine.
Hereditary coproporphyria (HCP)
It is caused by an AD hereditary defect of coproporphyrinogen oxidase.
Symptoms include neuropsychiatric problems, photosensitivity, rarely abdominal pain, but completely asymptomatic forms are also common. In the acute stage, there are increased levels of ALA, PBG, and coproporphyrin in urine (this is also detectable in the stool).
Porphyria variegata (VP)
This is an AD inherited defect of protoporphyrinogen oxidase.
Symptoms include abdominal pain, neuropsychiatric problems, and, in some patients, skin symptoms (photosensitivity). High levels of ALA, PBG, and coproporphyrin are present in urine and there is increased excretion of protoporphyrin and coproporphyrin in feces.
Diagnosis
This is done by the detection of accumulating porphyrins in body fluids, urine, and feces (specific fluorescence).
Note: Porphyrinuria and ALA accumulation may also be a symptom of lead poisoning (since lead inhibits ALA dehydratase and ferrochelatase), resulting in anemia and ATP deficiency.
Treatment of porphyria
Patients should pay stick to an appropriate lifestyle: proper nutrition with sufficient vitamins, avoiding alcohol and garlic (contains enzymes that worsen the symptoms of porphyria), and minimizing sunlight and UV radiation exposure. Blood transfusions and heme injections are used for symptomatic treatment.
References
Related articles
External links
Source
MASOPUST, Jaroslav a Richard PRŮŠA. Patobiochemie metabolických drah. 2. vydání. Univerzita Karlova, 2004. 208 s. s. 118–119.
Citations
Literature
- MURRAY, Robert, Daryl GRANNER a Peter MAYES, et al. Harperova biochemie. 4. české vydání. Jinočany : Nakladatelství H+H, 2002. 872 s. s. 354-360. ISBN 80-7319-013-3.
- LEDVINA, Miroslav, Alena STOKLASOVÁ a Jaroslav CERMAN. Biochemie pro studující medicíny : II.díl. 2. vydání. Praha : Nakladatelství Karolinum, 2009. s. 336-341. ISBN 978-80-246-1415-1.
- MASOPUST, Jaroslav a Richard PRŮŠA. Patobiochemie metabolických drah. 1. vydání. Praha : Univerzita Karlova, 2. lékařská fakulta, 1999. 182 s. s. 104-110. ISBN 80-238-4589-6.
- KALOUSOVÁ, Marta, et al. Patobiochemie ve schématech. 1. vydání. Praha : Grada Publishing a.s, 2006. 264 s. s. 51-57. ISBN 80-247-1522-8.

{kind=link}